|
НАУЧНЫЕ РЕЗУЛЬТАТЫ 2009 ГОДА
[Публикации 2009 года]
1. ОБЩИЕ ПРОБЛЕМЫ КВАНТОВОЙ ТЕОРИИ МОЛЕКУЛ [наверх]
1.1. Детально рассмотрены базовые положения, связанные с постановкой квантовых задач в теории строения и свойств молекулярных объектов. Акцентировано внимание на том, что формулировка проблемы в форме общего уравнения Шредингера с учётом лишь фундаментальных (кулоновских) взаимодействий между электронами и ядрами является недостаточной и не приводит к практически пригодным результатам для молекул, содержащих более 6-10 атомов. Необходима дополнительная информация, которая в принципе не может быть получена только средствами квантовой механики и свидетельствует об её ограниченности. Используя только понятия и аппарат квантовой механики, невозможно априори получить данные о геометрической структуре объекта, необходимые для доопределения проблемы.
Квантовые задачи для молекулы должны ставиться так, чтобы с самого начала вводились ограничения на возможные структурные формы анализируемых молекулярных систем. Обоснование соответствующих подходов и форм гамильтонианов наиболее просто получить, исходя из интегрального выражения для энергии объекта.
Предложено общее матричное уравнение для объединенных электронно-ядерных состояний. Это уравнение позволяет единым образом описывать переходы, приводящие как к ИК, так и к УФ (с учётом колебательной структуры) спектрам. Этот подход опирается на новый электронно-ядерный гамильтониан, впервые учитывающий квантованность слагаемых для электронно-ядерных взаимодействий. Гамильтониан предусматривает разделение электронных и ядерных движений и дополнительные условия, связанные с фиксацией определенного геометрического строения системы. Уровни энергии и волновые функции электронно-ядерной задачи зависят от введённых параметров потенциала для движений ядер. Тем самым создаются условия для согласования решения полной обратной задачи об электронно-колебательных уровнях энергии молекулы как для основных, так и для возбуждённых состояний.
Показано, что практически пригодные методы решения квантовых задач получаются только при сочетании операторного (дифференциальные уравнения) и матричного формализмов, причём они не эквивалентны, а являются взаимодополняющими (по Н.Бору). Получены матричные уравнения, адекватные уравнениям Шредингера, в условиях избыточных базисов и при осцилляторных базисных функциях, не отвечающих стационарным состояниям квантовой задачи. Указаны методы формирования матриц и способы вычисления матричных элементов. Показано, что некоторые общие закономерности в колебательных спектрах высших порядков и в электронно-колебательных спектрах могут быть объяснены без привлечения понятий ангармонизма и изменений параметров потенциальной ямы для электронно-возбуждённого состояния по сравнению с основным. Созданы основы новой теории электронно-колебательных спектров молекул с единой системой параметров и с полным учётом фундаментальных электронно-ядерных взаимодействий.
1.2. Важнейший результат по фундаментальным исследованиям в квантовой механике молекул получен в области релятивистской квантовой теории молекулярных систем. Доказана теорема о представлении точного решения уравнения Дирака для электрона молекулы в виде ЛКАО, где роль атомных орбиталей играют атомные биспиноры. Тем самым метод МО ЛКАО в расчетах молекул с элементами всей таблицы Менделеева получает статус физико-теоретического обоснованного метода, адекватно отражающего структурные особенности электронной плотности молекул. Другой фундаментальный результат в релятивистской квантовой химии относится к установлению дополнительного двукратного вырождения решений уравнения Дирака для электрона в электрическом поле, связанного с вырождением спиновых состояний. Этот результат позволяет объединить теорию электронных релятивистских мультиплетов в системе многих электронов с теорией спиновых нерелятивистских мультиплетов в одну расчетную схему построения электронных конфигураций. При этом достигается автоматический переход от релятивистской схемы к нерелятивистской обнулением матричных элементов при переходе к бесконечному значению скорости света.
1.3. По проблемам вычислительной физики велась разработка метода вычисления матричных элементов квантовой химии от базисных спинорных функций. Эта работа подготовляет этап по созданию комплекса программ «БЕССЕЛИАН», в котором интегральный модуль вычисляет матричные элементы от базисных функций с атомной асимптотикой (а не гауссовой, как в комплексе «ГАУССИАН»).
Разработан метод вычисления матричных элементов от произвольной функции R(x) с функциями Морзе с помощью представления матричного элемента M=<n|R(x)|m> в виде операторного произведения M=R(d/ds)J(s)|s=0, где вклады от R(x) и функций Морзе выступают в разделенном (факторизованном) виде. Получено выражение базисного интеграла J(s)=<n|exp(-sx)|m> в виде двойного ряда Аппеля F2(1,1). Показано, что этот ряд допускает представление в виде симметричного по числам n, m однократного гипергеометрического ряда 3F2(1) полиномиального типа от единичного аргумента.
2. ТЕОРИЯ ФОТОХИМИЧЕСКИХ РЕАКЦИЙ [наверх]
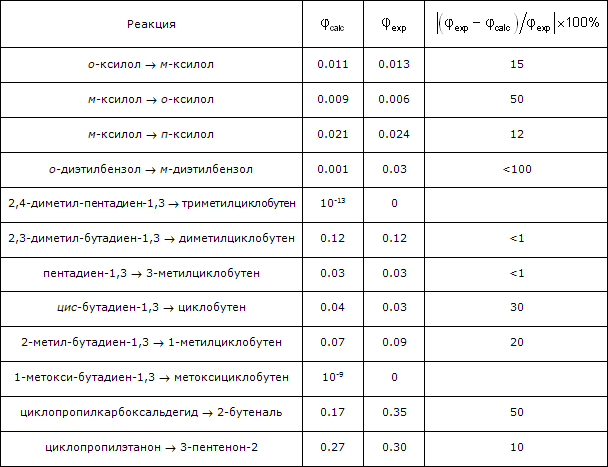
2.1. Исследована возможность построения полуэмпирического метода моделирования фотохимических процессов и расчета квантовых выходов реакций. Предложен полуэмпирический подход, опирающийся на основные физические принципы и адекватный логике построения всех работоспособных физических теорий в спектроскопии и квантовой химии. Результаты выполненных расчетов фотоизомеризации ряда молекул (12 реакций) показали работоспособность развиваемого метода – обеспечивается количественное прогнозирование квантовых выходов реакций на хорошем уровне согласия с имеющимися экспериментальными данными. Для реально наблюдаемых реакций с квантовыми выходами в пределах 0.01–1 отклонение прогнозируемых значений от экспериментальных составило 10–50 %. В тех случаях, когда по экспериментальным данным реакции не идут, рассчитанные значения квантовых выходов пренебрежимо малы – менее 10-9. Количественно верно предсказывается и соотношение величин квантовых выходов рассмотренных фотопревращений молекул, включая и случай аналогичных фотохимических процессов с качественно (на порядки величин) различающимися значениями квантовых выходов. Установлено, что при выполнении условия относительной малости вероятностей оптических переходов по сравнению с частотой квантовых биений количественные оценки квантовых выходов с удовлетворительной степенью точности могут быть получены непосредственно по величинам вероятностей переходов без проведения полного расчета кинетики фотопревращения. Выявлена причина наблюдающегося в эксперименте существенного различия квантовых выходов однотипных реакций для ряда молекул, при разных заместителях – изменение форм колебаний и/или изменение величины структурной перестройки, когда заместитель непосредственно в ней участвует. Показана высокая чувствительность интегральных характеристик фотохимических реакций (квантовых выходов) к конформации участвующих в процессе молекул.
2.2. В качестве необходимого при описании бимолекулярных реакций способа определения свойств димера (промежуточного комплекса) использовались квантово-химические расчёты, с помощью которых можно подтвердить существование самого димера и оценить его геометрические характеристики и характеристики потенциальной ямы, а при наличии нескольких вариантов стабильной конфигурации выбрать наиболее выгодный по энергии. Проведены квантово-химические расчёты устойчивых взаимных расположений простых органических молекул – продуктов реальных фотохимических реакций разложения.
2.3. Вычисление интеграла перекрывания колебательных волновых функций двух изомерных форм, использующее выражение для связи нормальных координат Q1 и Q2 в виде обобщённого соотношения Душинского сопряжено с рядом проблем, вызванных необходимостью перезадания некоторых угловых координат, приведением к одному типу разных по сути естественных координат и т.п. Задание единой для обоих изомеров центрально-силовой системы естественных координат помогает преодолеть указанные трудности. В этом случае условия обратимости выполняются автоматически, а отсутствие в такой системе всех типов координат, кроме координат изменения длин связей, снимает проблему контроля за угловыми и неплоскими координатами.
3. ФИЗИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ МОЛЕКУЛ [наверх]
3.1. Развит ранее предложенный новый подход к теоретическому описанию спектров КР как результата резонансного поглощения энергии электромагнитного поля, описываемого модулированным сигналом. Показано, что на этой основе можно ввести обобщенный оператор возмущения в форме матрицы, одновременно учитывающей как поглощение, так и рассеяние. Предложен способ формирования симметричной энергетической матрицы, описывающей все состояния молекулы и их взаимодействие. Получающийся простой вид электронно-колебательной функции позволяет существенно упростить вычисления матричных элементов для оптических переходов между уровнями энергии молекул.
3.2. В теории расчета термических средних, которая является фундаментальной задачей в теории рассеяния частиц на молекулярных структурах (электронография, нейтронография, рассеяние рентгеновых лучей) разработан новый метод. Вкратце теория метода состоит в том, что рассеяние электронов происходит не на точечных ядрах молекулы, а на ядерных плотностях, обусловленных движением ядер при больцмановском распределении ансамбля молекул в газе по колебательным состояниям. Тогда измеряемые косвенно в эксперименте средние расстояния между ядрами в молекуле следует интерпретировать как термические средние расстояния. Плотность состояний молекулы удовлетворяет уравнению Блоха, напоминающему временное уравнение Шрёдингера с заменой переменной времени на обратную абсолютную температуру. Переведя это уравнение к интегральному виду, удается дать аналитические способы вычисления термических средних межатомных расстояний, а также вычислить термодинамические функции молекулярного газа с учетом ангармонических поправок в колебательном потенциале.
3.3. В последнее десятилетие квантово-химические (QM) методы расчета спектров ЯМР на основе теории функционала плотности (DFT) в приближении GIAO получили широкое распространение. Рядом авторов высказывается мнение, что эти методы стали почти рутинными и предлагается использовать их для выявления наиболее правдоподобной структурной гипотезы. Связанные с квантовыми расчётами трудности заставляют обратиться к другим подходам. Были существенно усовершенствованы эмпирические методы предсказания спектров ЯМР с помощью алгоритмов, основанных на фрагментном подходе (использование кода HOSE), аддитивных правилах и использовании искусственных нейронных сетей. Эмпирические методы
-
полностью автоматизированы
-
не имеют ограничений по размерам молекул
-
обладают высокой скоростью (200-400 спектров в секунду)
-
весьма высокой точностью (средняя абсолютная ошибка 1.5-1.8 мд.).
Очевидно, что QM методы как рутинные методы расчета неконкурентоспособны по пунктам (1)-(3), однако они позволяют предсказывать спектры молекул, содержащих ранее неизвестные фрагменты (в таких случаях эмпирические методы производят интерполяцию). В связи с этим необходимо оценить возможную точность QM расчетов в сравнении с точностью эмпирических методов. До настоящего времени результаты расчетов спектров ЯМР 13С эмпирическими и неэмпирическими методами на большом множестве молекул не сравнивались. Эта задача была решена нами в 2009 г.
С этой целью были отобраны в литературе более 100 работ, в которых приведены экспериментальные и рассчитанные QM методами химические сдвиги в спектрах ЯМР 13С органических молекул. Структуры этих молекул вместе с их экспериментальными и расчетными спектрами были введены в экспертную систему Structure Elucidator и создана база данных, включающая 205 молекул. Для всех молекул были рассчитаны спектры ЯМР 13С тремя эмпирическими методами, и с помощью специально разработанной программы произведена полная статистическая обработка массивов данных. Статистический анализ показал, что в принципе, при удачном выборе вида функционалов DFT и базисных наборов, QM методы могут обеспечить точность, близкую к точности эмпирических методов. Однако для данного тестового множества средняя точность эмпирических методов оказалась в 1.5-2 раза выше точности квантовых. Предложена методология совместного применения эмпирических и неэмпирических методов для предсказания ЯМР спектров новых органических соединений. Ожидается, что такой подход будет наиболее экономным и эффективным.
3.4. В ангармоничном приближении рассчитаны интенсивности фундаментальных, обертонных и составных полос поглощения для 11 непредельных углеводородов, 17 азот- и кислородсодержащих и 12 серосодержащих органических соединений. Первые и вторые производные от дипольного момента молекулы вычислялись квантово-химически в ab initio приближении с использованием МР2/6-31G(1d) базиса. Получено, что для изученных соединений средний вклад обертонов и составных частот в поглощение в области от 100 до 4000 см-1 составляет ~10%. При этом большая часть этого вклада (в среднем 80%) приходится на область 1600–2800 см-1, в которой нет фундаментальных переходов. Проведенный расчёт хорошо передаёт положение и интенсивности фундаментальных, обертонных и составных полос поглощения и может применяться для безэталонного спектрального анализа данных соединений по их обертонным спектрам.
С использованием B3LYP/6-311G(3df,3pd) базиса произведено квантово-химическое вычисление параметров потенциальной и электрооптической функции 32-х гетероатомных (азот-, кислород- и серосодержащих) соединений. Получено, что после небольшой коррекции потенциальной функции с помощью единых значений масштабирующих множителей удается достичь удовлетворительного согласия вычисленных и экспериментальных частот и абсолютных интенсивностей фундаментальных полос поглощения. Среднее и среднеквадратичное отклонения вычисленных частот от экспериментальных не превышает полуширины их полос поглощения. Результаты указывают на возможность использования таких вычислений для безэталонного спектрального анализа газовых смесей.
4. МЕХАНОХИМИЯ [наверх]
4.1. В области общей теории механохимии в отчетном году основная работа была связана с исследованием процессов возбуждения колебательных движений в протяженных молекулярных системах ударом. Результаты этой работы имеют отношение к прояснению условий инициации реакций и изоморфных превращений в молекулах и кристаллах при столкновениях молекул и при бомбардировке поверхностей таких систем быстрыми частицами. Полученные результаты позволяют дать физическую интерпретацию ряду известных химических и биофизических явлений. В частности, показано, что в анизотропных молекулярных средах имеются определенные каналы, по которым могут распространяться волны колебательных возбуждений, способные вызвать во внутренних реакционных центрах структурные перестройки, которые обычно возбуждаются тепловым движением или электромагнитным излучением. С другой стороны, существуют такие системы, которые совершенно неспособны реагировать на внешние случайные удары. Такие условия создаются, например, ферментами на время протекания катализируемых ими реакций. Этот биофизический факт, получивший теперь физическое обоснование, может иметь прямое отношение к решению проблемы эволюции живого вещества, поскольку стандартизация сценариев протекания ферментативных реакций способствует стабилизации живого вещества в самых неблагоприятных условиях, в которых существует вся биосфера Земли.
4.2. Обращено внимание на то, что одним из факторов, стимулирующих изоморфные превращения в минералах, могут быть возникающие в результате землетрясений внешние импульсные механические воздействия на кристаллическую структуру минералов. При подходящих условиях такие воздействия могут привести к фокусировке энергии при возмущении ряда участков поверхности кристалла и стимулировать прохождение химической реакции. Показано, что механическая прочность минерала определяется не особенностями химических связей, а диссипацией вводимой энергии по всему пространству объекта. Предложен метод расчёта соответствующих эффектов, пригодный для проведения компьютерных экспериментов.
5. ЧАСТНЫЕ ЗАДАЧИ [наверх]
5.1. Совместно с ИСАН РАН на базе Центра по фемтооптике, имеющего источники перестраиваемого лазерного излучения фемтосекундной длительности, начаты работы по исследованию возможностей фемтосекундной лазерной спектроскопии в аналитической химии. Создан лазерный одноканальный фемтосекундный ИК спектрометр для изучения быстрых процессов в газовой фазе. Используя технику возбуждения-зондирования (pump\probe), исследовали ИК спектры возбуждения молекул (CF3)2CCO, Fe(CO)5 и Cr(CO)6 и их эволюцию во времени.
Во всех исследованных молекулах обнаружены быстропротекающие фемто и пикосекундные процессы, измерены их характеристики. Экспериментально наблюдали несколько процессов релаксации. Быструю кинетику с характерными временами 400-500 фс наблюдали для молекул Fe(CO)5 и Cr(CO)6, более медленная в виде спадающей по экспоненте кривой с характерным временем 5 пс наблюдалась для молекулы (CF3)2CCO. На основе полученных данных определены времена релаксации возбуждаемых переходов пропорциональные соответствующим вероятностям переходов. Анализ данных позволяет для модельных молекул найти условия повышения селективности анализа молекулярных смесей и уменьшения или исключения влияния матриц на аналитический сигнал при использовании фс техники.
Показана возможность увеличения селективности определения молекул, используя разницу в экспериментальных временах релаксации и технику временной селекции. Во всех молекулах наблюдали и медленные процессы, идущие в наносекундном диапазоне, которые могут ограничивать чувствительность и селективность. Продемонстрирована возможность определения концентраций с использованием экспериментальных данных Практические применения техники расширяются при работе с двумя независимыми каналами. Для этой цели проводится усовершенствование лазерного спектрометра, создание двухканального варианта спектрометра.
5.2. Определяющим признаком феномена жизни является связанная с закономерностями эффекта репликации направления передачи информации от прошлого к будущему. Показано, что эволюционный выбор направления, приводящего к максимальной плотности передачи информации, определяется соотношениями вероятностей рождения и смерти воспроизводимых структур. Необходимое для повышения эффекта самоорганизации уменьшение потока негоэнтропии достигается при увеличении разнообразия воспроизводимых объектов. Задаваемое генетическим кодом выделенное направление эволюции и свойство множественности объектов биосферы находятся во взаимодополняющем соотношении.
5.3. Предложен метод расчёта электронных и колебательных уровней энергии нанообъектов с периодической внутренней структурой. Метод позволяет в хорошем приближении свести общую задачу к ряду задач меньшего порядка, отвечающих повторяющейся совокупности атомов. Дальнейшее уточнение делается методом теории возмущений.
| 
